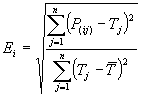| GeneXproTools 4.0 implements the Root Relative Squared
Error (RRSE) fitness function both with and without parsimony
pressure. The version with
parsimony pressure puts a little pressure on the size of the
evolving solutions, allowing the discovery of more compact models. The RRSE fitness function of GeneXproTools is, as expected, based on the standard root relative squared error, which, on its turn, is based on the absolute error. The relative squared error is relative to what it would have been if a simple predictor had been used. More specifically, this simple predictor is just the average of the actual values. Thus, the relative squared error takes the total squared error and normalizes it by dividing by the total squared error of the simple predictor. By taking the square root of the relative squared error one reduces the error to the same dimensions as the quantity being predicted. Mathematically, the root relative squared error Ei of an individual program i is evaluated by the equation:
where P(ij) is the value predicted by
the individual program i for fitness case j (out of n
fitness cases or sample cases); Tj is the target value for fitness
case j; and
For a perfect fit, the numerator is equal to 0 and Ei
= 0. So, the RRSE index ranges from 0 to infinity, with 0
corresponding to the ideal.
which obviously ranges from 0 to 1000, with 1000 corresponding to the ideal.
where Si is the size of the program, Smax and Smin represent, respectively, maximum and minimum program sizes and are evaluated by the formulas: Smax = G (h + t) Smin = G where G is the number of genes, and h and t are the head and tail sizes (note that, for simplicity, the linking function was not taken into account). Thus, when rfi = rfmax and Si = Smin (highly improbable, though, as this can only happen for very simple functions as this means that all the sub-ETs are composed of just one node), fppi = fppmax, with fppmax evaluated by the formula:
|